Reconstructing the infrared spectrum of a peptide from representative conformers of the full canonical ensemble
Abstract
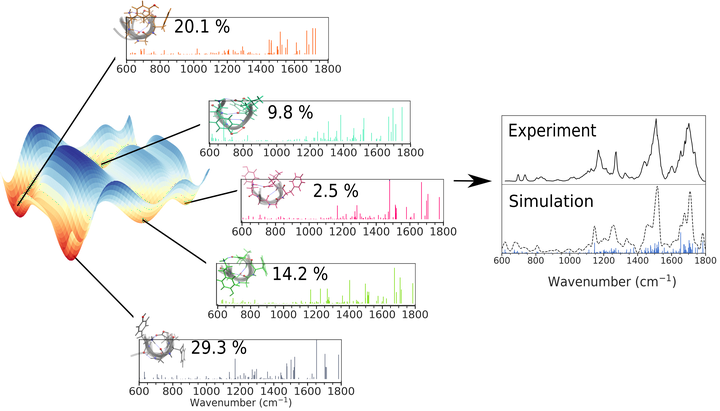
Leucine enkephalin (LeuEnk), a biologically active endogenous opioid pentapeptide, has been under intense investigation because it is small enough to allow efficient use of sophisticated computational methods and large enough to provide insights into low-lying minima of its conformational space. Here, we reproduce and interpret experimental infrared (IR) spectra of this model peptide in gas phase using a combination of replica-exchange molecular dynamics simulations, machine learning, and ab initio calculations. In particular, we evaluate the possibility of averaging representative structural contributions to obtain an accurate computed spectrum that accounts for the corresponding canonical ensemble of the real experimental situation. Representative conformers are identified by partitioning the conformational phase space into subensembles of similar conformers. The IR contribution of each representative conformer is calculated from ab initio and weighted according to the population of each cluster. Convergence of the averaged IR signal is rationalized by merging contributions in a hierarchical clustering and the comparison to IR multiple photon dissociation experiments. The improvements achieved by decomposing clusters containing similar conformations into even smaller subensembles is strong evidence that a thorough assessment of the conformational landscape and the associated hydrogen bonding is a prerequisite for deciphering important fingerprints in experimental spectroscopic data.
Gregor Vonbun-Feldbauer
Projektleiter & MLE-Koordinator
Computergestützte Materialwissenschaften, Atomistische Simulation & Modellierung, Multiskalenansätze, Maschinelles Lernen und datengetriebene Ansätze für Materialmodellierung & -design, Hybrid- und Mehrkomponentensysteme, Ober- und Grenzflächenphysik
Christian Feiler
Postdoc & MLE-Koordinator
Atomistische Simulation & Modellierung, Entwicklung von Quantitativen Struktur-Eigenschafts-Beziehungsmodellen
Robert Meißner
Professor
Molekulardynamische Simulationen von Grenzflächen, Berechnung freier Energien biomolekularer und elektrochemischer Systeme, atomistische Betrachtung der Magnesiumkorrosion, Entwicklung datengetriebener Modelle zur Indentifikation von (umweltfreundlichen) Degradationsmodulatoren